Screenshots
-- Screenshot 1 --
-- Screenshot 2 --
-- Screenshot 3 --
-- Screenshot 4 --
-- Screenshot 5 --
-- Screenshot 6 --
-- Screenshot 7 --
-- Screenshot 8 --
-- Screenshot 9 --
-- Screenshot 10--
-- Screenshot 11--
-- Screenshot 12--
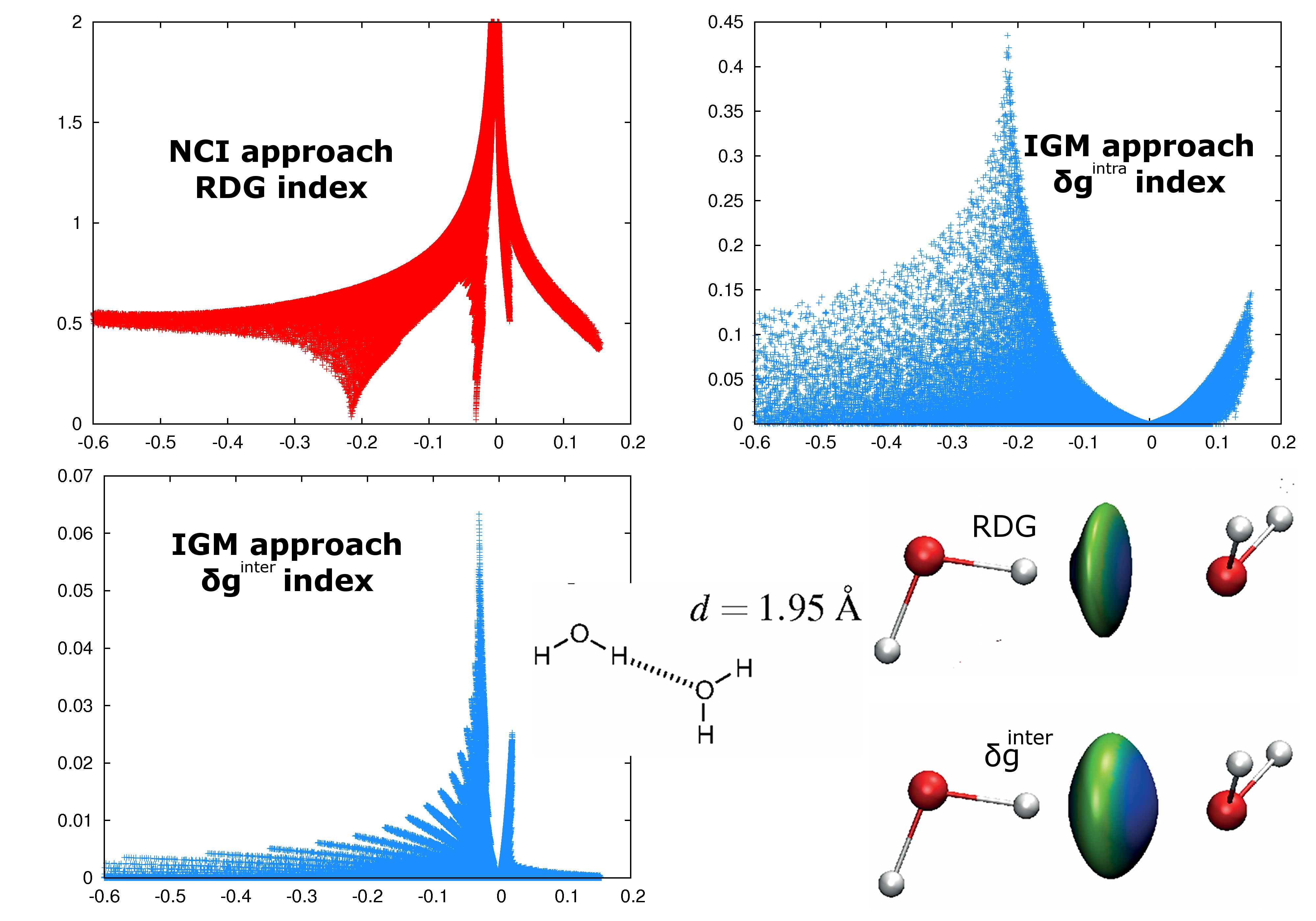
An attractive feature of the IGM methodology is to provide an automatic intra/inter uncoupling scheme that extracts the signature of intra- and intermolecular interactions for drawing the corresponding 3D iso-surface representations in real space with software like VMD (available with promolecular ED and ED coming from QM calculations).
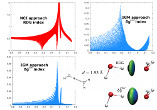
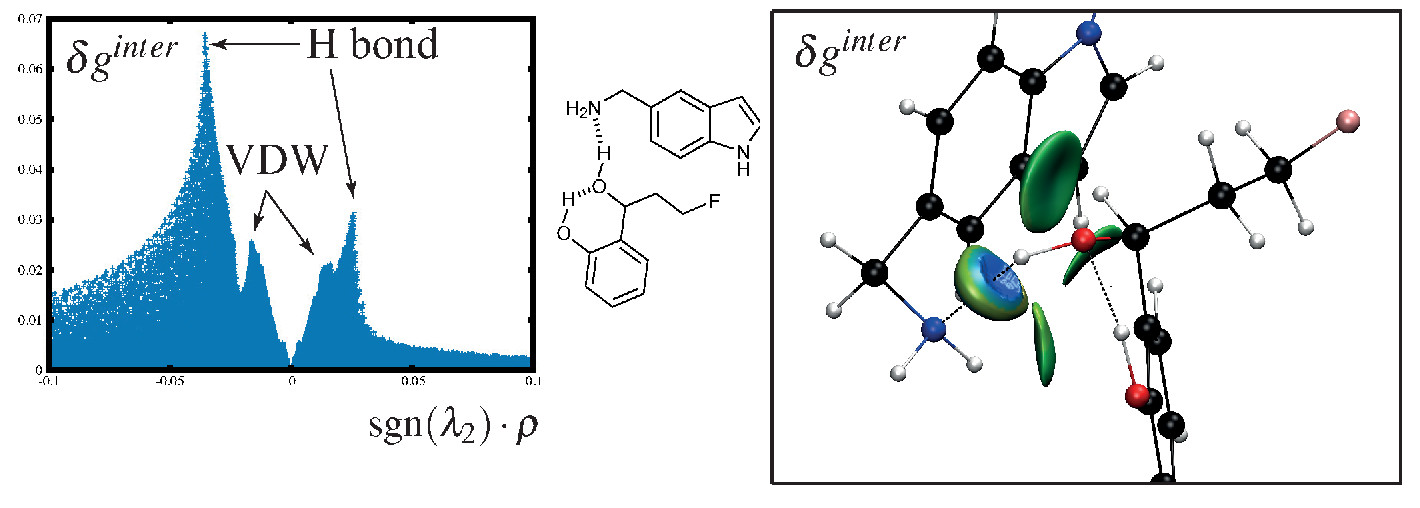
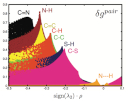
The δginter descriptor is not dimensionless, linking its value to the familiar concept of interaction strength. In particular, the peak height is related to the interaction energy, with for instance hydrogen-bonding displaying larger peaks than vdW.
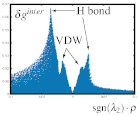
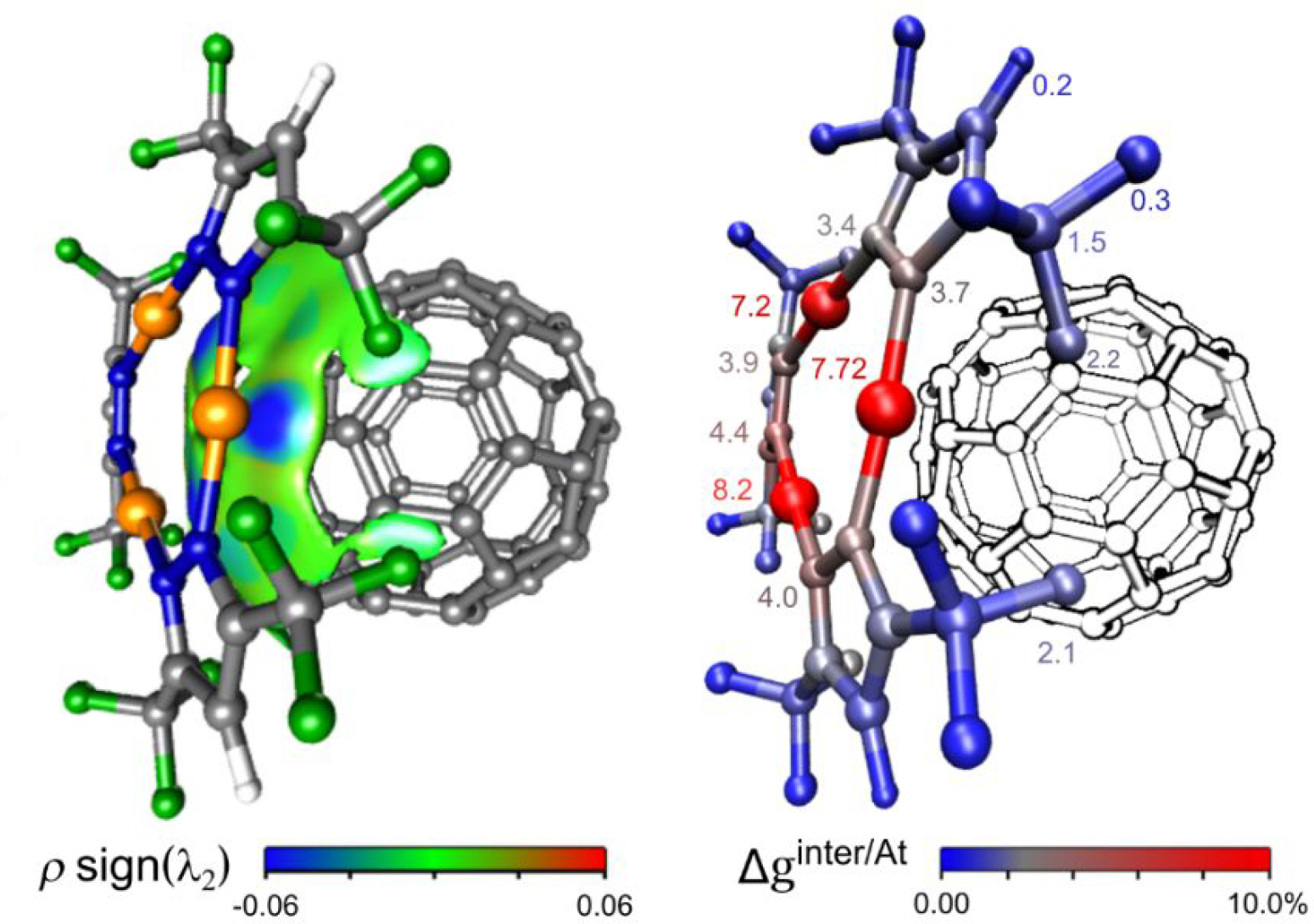
In the “promolecular” mode, a compelling feature of the IGMPlot program is to carry out an atomic decomposition scheme in order to estimate the influence of a given atom in the intermolecular region between two user-selected fragments. This complementary analysis brings a quantitative side to the IGM approach.
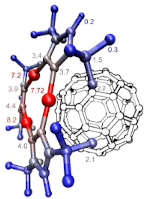
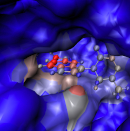
The IGM-δ approach is particularly suited to studies of host-ligand complexes using promolecular electron density. In tandem with δginter isosurfaces, the atomic scheme decomposition δgAt helps identifying ligand and protein hot-spots.
The IGM-δginter approach allows for distinguishing between
weak interactions in multimers that would be hardly separable otherwise.
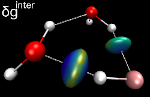
User-defined fragments can be sub-units of a single molecule !
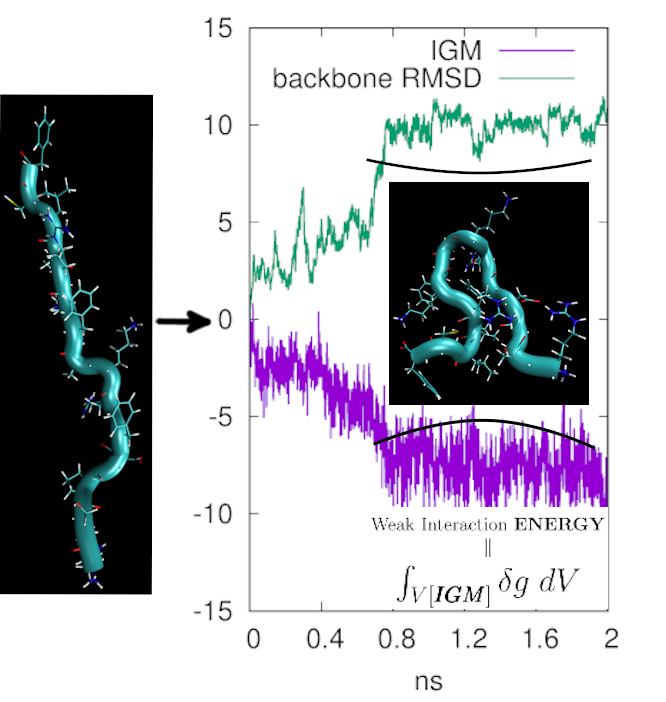
Hence, intramolecular π-π stacking can be described and quantified,
even at the quantum-mechanical level.
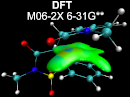
From a wave function, the IGM-IBSI index is very efficient to internally probe the intrinsic strength of a given atom pair, over a wide range (non-covalent to covalent, transition metal bonding, agostic interactions, ...).
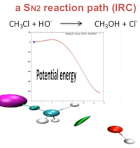
IGM paves the way for the targeted mechanistic exploration along reaction pathways through QM calculations.
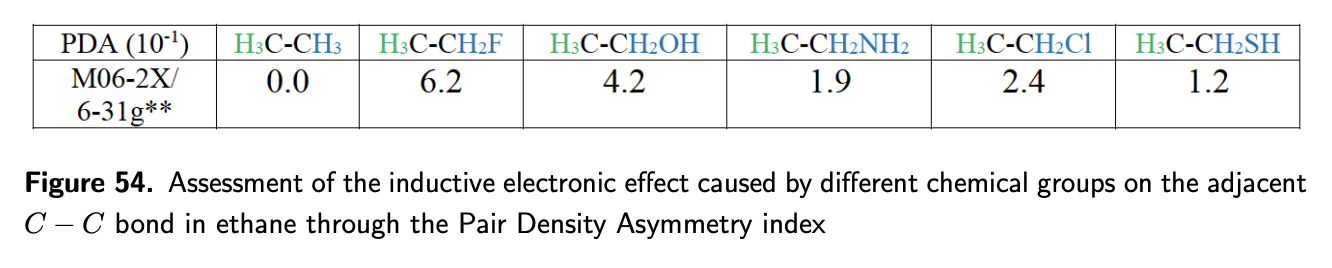
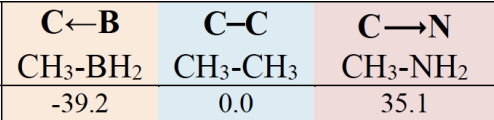
In order to provide users with a simple tool to assess inductive effects on specific bonds in
molecules, the new Pair Density Asymmetry (PDA) index has been devised. The PDA index
gives a measure of the ED asymmetry in between the two atoms and the direction of the
asymmetry.
An integration scheme has been implemented to relate the δg signature to the interaction energy between two fragments or either to the intramolecular weak interaction energy.

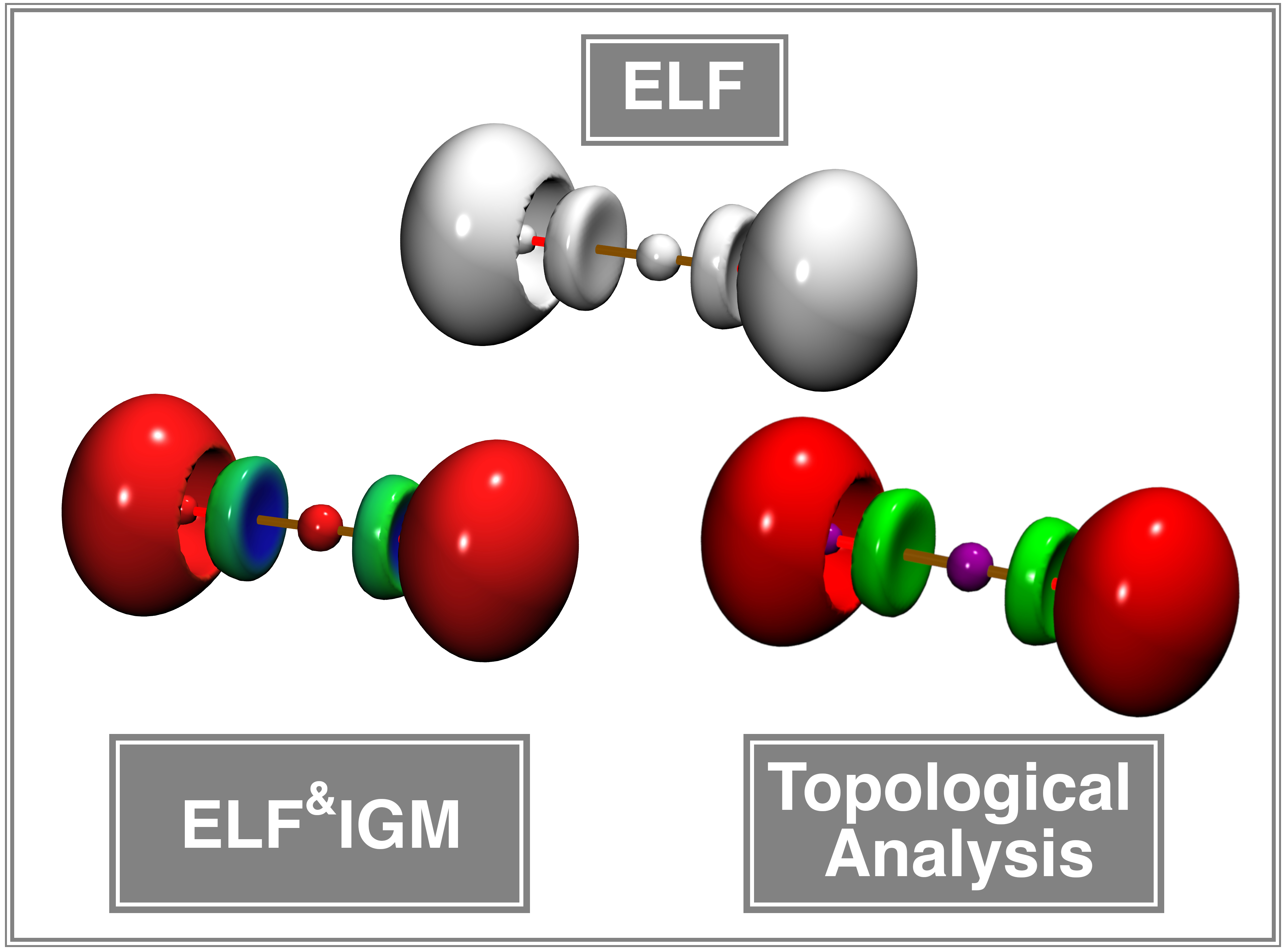
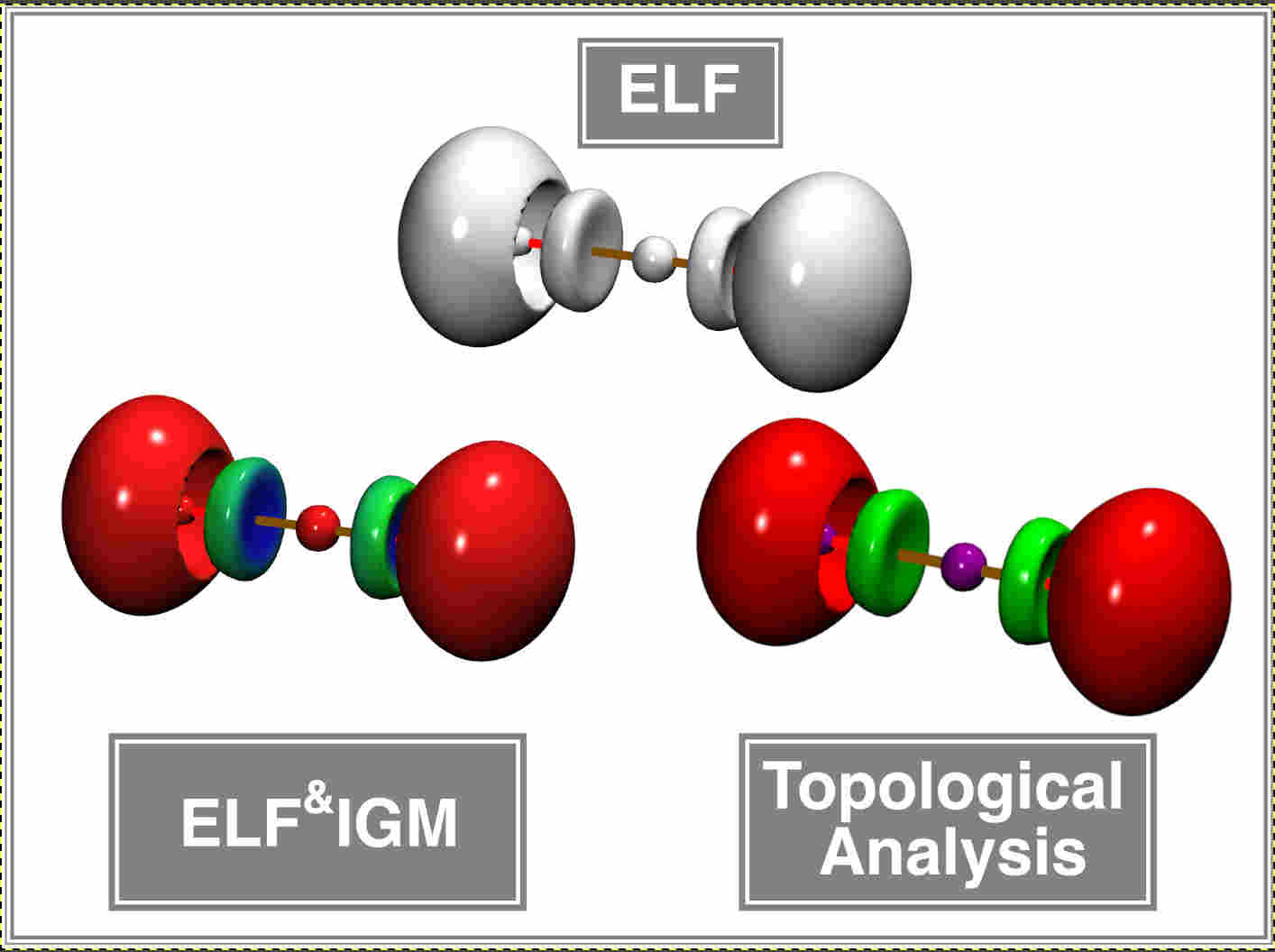
The ELF&IGM approach
determine ELF basin characteristics while achieving
computational efficiency gains of approximately two orders of magnitude compared to full
topological analysis. This makes it a pragmatic and accessible alternative for high-throughput
ELF studies while preserving essential qualitative insights.
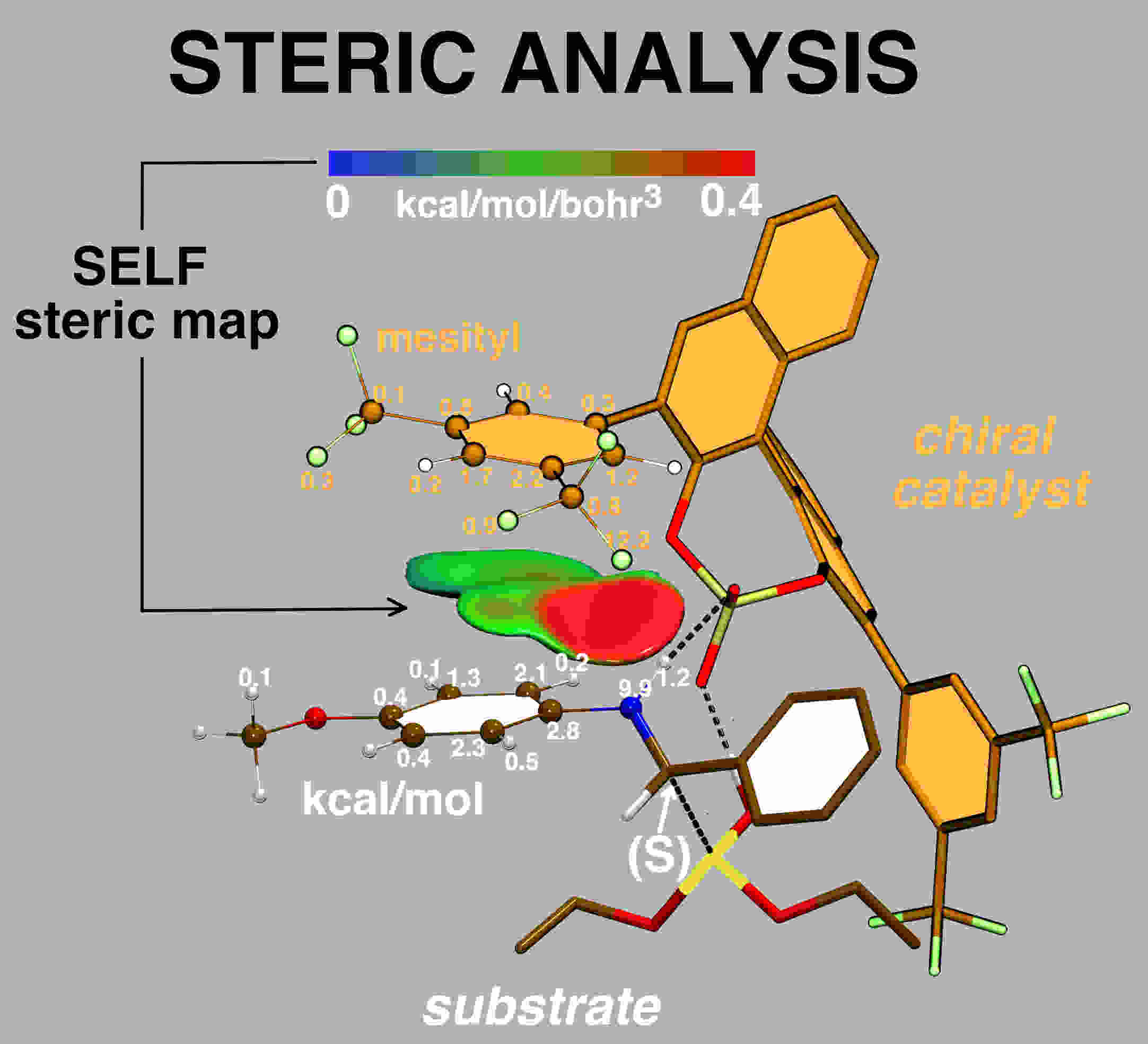
A new approach, SELF: so-called ”Steric Exclusion Localization Function” has been devised to provide
users with a spatial characterization of the steric effect together with a quantification and atomic
decomposition.
#